
目录
各类生信文件官方解读手册
http://genome.ucsc.edu/FAQ/FAQformat.html
经典文件格式解读
1. fasta
fasta文件:核酸和氨基酸序列文件
两行。
第一行:以">"开头,为序列名称行,包括各类信息,可以有序列长度等等。
第二行:序列行。(有多行和单行之分)
统计序列信息
#统计序列条数 grep -c ">" ./test.fasta #统计序列总长度 #若序列行为单行 awk 'BEGIN{number=0;len=0} {if($0~/^>/){number+=1} else{len+=length($0)}} END{printf "%s%d\t%s%d\n", "Total number: ",number,"Total length: ",len }' ./test.fa #这个只能统计累积和最高为2,147,483,647。因此存在bug。 #若序列行为多行,请先多行转单行,再统计 awk '/^>/&&NR>1{print "";}{printf "%s", /^>/?$0"\n":$0}' ./test.fa > ./test.fa.tmp 当然,可以写python/perl脚本进行更加详细的统计
2. fastq
fastq文件:测序结果文件
四行。
第一行:以"@“或者”>“开头,为序列名称行,包括各类信息,可以有测序信息、序列长度等等。
第二行:序列行。(单行)
第三行:”+"或者重复第一行
第三行:序列质量行。(单行),长度与第二行一致。
统计序列信息
#统计序列条数 grep -c "@" ./test.fq grep -c ">" ./test.fq #统计序列总长度 #直接对fastq文件进行统计 awk 'BEGIN{number=0;len=0} {if(NR%4 == 1){number+=1}} {if(NR%4 == 2){len+=length($0)}} END{printf "%s%d\t%s%d\n", "Total number: ",number,"Total length: ",len }' ./test.fq #这个只能统计累积和最高为2,147,483,647。因此存在bug。 #fastq格式转fasta格式 awk '{if(NR%4 == 1){print ">"substr($0,2)}} {if(NR%4 == 2){print}}' test.fq > test.fa 3. sam与bam
bam文件:sam的二进制文件,不能直接查看。需要使用软件samtools
sam文件:比对结果文件,通常有11个主列+可选列,不同软件比对后的结果文件在可选列间存在差异。
比对软件:bowtie,hisat,bwa,hifiasm等
第1列:read编号
第2列:数值,也称Flag/标签值,由2的n次方加和。例如99,147,83,163等
99=1+2+32+64 147=1+2+16+128
1代表这个序列采用的是PE双端测序
2代表这个序列和参考序列完全匹配,没有插入缺失
4代表这个序列没有比对到参考序列上
8代表这条序列的配对序列也没有比对到参考序列上。如果这条是R1,它对应的R2端没有比对上
16代表这个序列比对到参考序列的负链上
32代表这个序列对应的另一端序列比对到参考序列的负链上
64代表这个序列是R1端序列
128代表这个序列是R2端序列
256代表这个序列不是主要的比对,一条序列可能比对到了多个位置,只有一个是首要的比对位置
512代表这个序列在QC时失败了,被过滤不掉了
1024代表这个序列是PCR重复序列
2048代表这个序列是补充的比对
第3-4列:参考序列编号,比对到参考序列上的位置。代表这条read比对到4号染色体的这个位置
第5列:数值,比对的质量分数,越高说明该read比对到参考基因组上的位置越唯一
第6列:CIGAR值,碱基匹配上的碱基数。
M:match/mismatch;I:insert;D:deletion;N:skipped(跳过这段区域)Ssoft clipping(被剪切的序列存在于序列中)Hhard clipping(被剪切的序列不存在于序列中)Ppadding(填充)。135M:代表135个碱基在比对时完全比对;3S6M1P1I4M:代表前三个碱基被剪切去除了,然后6个比对上了,然后打开了一个缺口,有一个碱基插入,最后是4个比对上了,是按照顺序的。
第7列:mate reference sequence name,实际上就是mate比对到的参考序列编号。若是没有mate,则是*。若是"=",则代表与此条read和mate比对到相同的参考序列上。
第8列:mate position,mate比对到参考序列上的第一个碱基位置。若无mate,则为0
第9列:insert size
第10列:read序列。如果是比对到负链上则对read进行了reverse completed(反向互补)
第11列:read序列的质量行
第12行之后,是可选列。有NM-tag,XX-tag,XM-tag,XR-tag,XG-tag以及自定义列等
sam或bam文件查看
less test.sam samtools view test.sam | less samtools view test.bam | less #可以搭配grep,awk,sed等命令进行筛选查看 sam,bam,fasta,fastq文件转换
#sam转bam samtools view -@ 16 -b -S test.sam -o test.bam #参数-@:线程数; -b:输出格式为BAM -S:自动检测输入格式 #bam转sam samtools view test.bam -O SAM > test.sam #bam转fasta samtools view test.bam | awk '{print ">"$1"\n"$10}' > test.fa #bam转fasta samtools view test.bam | awk '{print ">"$1"\n"$10"\n+\n"$11"\n"}' > test.fq 解决samtools将sam文件转换为bam文件时多条重复错误的方法:Duplicate entry “1“ in sam header
这是sam文件Header部分出现问题,并不是比对结果出现问题
#安装bamutil
conda install -c bioconda bamutil
#sam转为bam
bam convert --in SRR.sam --out SRR_2.bam
#samtools 排序
samtools sort SRR_2.bam -o SRR_2_sorted.bam
#bam转换为无序bam (dedup 输入的文件必须是sorted的)
bam dedup --in SRR_2_sorted.bam --out ./SRR_3.bam
4. wig,bw,bdg文件
wiggle(简称wig)、bedgraph(简写bdg)文件以及bigwig(简写bw)只包含区域和区域的覆盖度信息,文件更小,用于可视化更方便,可以导入基因组浏览器(Genome Browser)中进行可视化,以查看reads在参考基因组各个区域的覆盖度并检测测序深度。这几个文件在ChIP-seq数据分析Call Peak阶段会生成,可以利用IGV或Tablet等软件进行可视化,方便查看组蛋白修饰的程度。
官网对三种类型文件的描述
wiggle(WIG):https://genome.ucsc.edu/goldenPath/help/wiggle.html
bedGraph:https://genome.ucsc.edu/goldenPath/help/bedgraph.html
bigWig:http://genome.ucsc.edu/goldenPath/help/bigWig.html
三种类型文件简述
wig文件,可以直接查看。
分为variableStep和fixedStep两种格式。variableStep用于区间变化的,fixedStep用于区间固定的。
variableStep文件以variableStep 开头,chrom染色体,可选参数span(默认span=1),用于指定每一行的位置区间。其他行两个值,第一个值chromStart,表示染色体上的起始位置,依据span的值,可知道染色体上的终止位置为chromStart+span。第二个值dataValue,表示值。

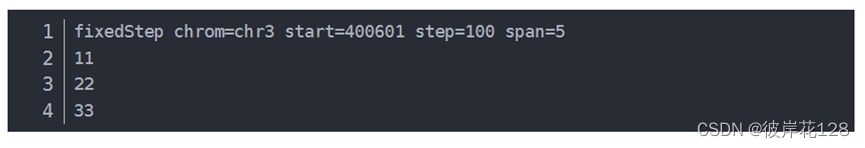
fixedStep文件以fixedStep开头,chrom染色体,start是起始固定的位置,step是每两个起始position之间的间隔,可选参数span(默认span=1),用于指定每一行的位置区间。其他行一个值dataValue,表示值。
bdg文件
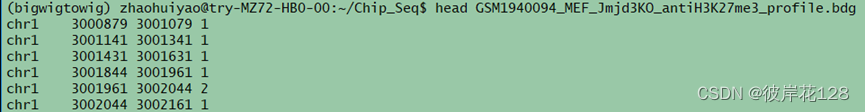
bed文件与wig文件的结合,更省空间和灵活,展示信息与wig类似。一般有四列,Chr、start、end和value,并且坐标是以0为起始左闭右开。
bigwig文件: wig文件和bdg文件的二进制压缩格式(不能直接查看)
wigToBigWig工具:wig文件转bw文件
wigToBigWig in.wig chrom.sizes out.bw
bedGraphToBigWig工具:bdg文件转bw文件
bedGraphToBigWig in.bedGraph chrom.sizes out.bw
bigWigToWig工具:bw文件转wig文件或bdg文件
bigWigToWig in.bw out.wig #这与wig文件的来源相关。如果从bdg创建了bigWig文件,则会将文件还原回bdg
bigWigToBedGraph工具:bw文件转bdg文件
bigWigToBedGraph in.bw out.bdg
#其他工具
bigWigSummary :从bigWig文件中提取摘要信息。
bigWigAverageOverBed :计算每个 bed 上可能有内含子的bigWig的平均得分。
bigWigInfo :打印出有关bigWig文件的信息。
conda create -n bigwigtowig
conda activate bigwigtowig
conda install -c bioconda ucsc-bigwigtowig #版本377
conda install -c bioconda ucsc-bigWigToBedGraph #版本377
conda install -c bioconda ucsc-wigtobigwig #版本447(若是出现报错,wigToBigWig: error while loading shared libraries: libssl.so.1.0.0: cannot open shared object file: No such file or directory。请使用低版本。conda install -c bioconda ucsc-wigtobigwig=366)
conda deactivate
cd /home/zhaohuiyao/Chip_Seq
#已知bw文件是由bdg文件创建
#查看bw文件

#转换文件
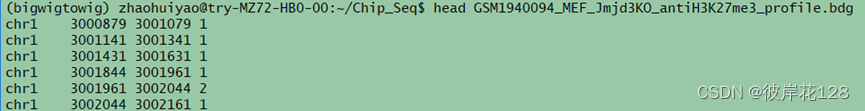
bigWigToBedGraph GSM_MEF_Jmjd3KO_antiH3K27me3_profile.bw GSM_MEF_Jmjd3KO_antiH3K27me3_profile.bdg
#查看bdg文件
Peak calling
5. narrow peaks format、broad peaks fotmat、gapped peaks format
3种peak格式:narrow peaks format、broad peaks fotmat、gapped peaks format
Narrow Peaks Format
该格式又称之为point-source peaks format,macs2默认输出就是这种格式,是一种BED6+4的格式,列数为10列,可以上传到UCSC浏览。该文件可以直接加载到UCSC基因组浏览器。
前四列分别代表chrom, chromStart, chromEnd, name, 用于描述peak区间和名称,注意bed格式中起始位置从0开始计数。
第五列score,在macs2的输出结果中为int(-10*log10qvalue),
第六列strand,,在macs2的输出结果中为 . ,
第七列signalvalue,通常使用fold_enrichment的值,
第八列pvalue,在macs2的输出结果中为-log10(pvalue),
第九列qvalue,在macs2的输出结果中为-log10(qvalue),
第十列peak,在macs2的输出结果中为peak的中心,即summit距离peak起始位置的距离。
该格式在BED12的基础上进行延伸,演变为BED12+3的格式,列数为15列。
今天的文章 各类生信文件解读(fasta,fastq,bam,sam,bed,bg,wig,bdg,gff3,gtf等)分享到此就结束了,感谢您的阅读。
版权声明:本文内容由互联网用户自发贡献,该文观点仅代表作者本人。本站仅提供信息存储空间服务,不拥有所有权,不承担相关法律责任。如发现本站有涉嫌侵权/违法违规的内容, 请发送邮件至 举报,一经查实,本站将立刻删除。
如需转载请保留出处:https://bianchenghao.cn/bian-cheng-ji-chu/103485.html